 |
Proteine stehen im Mittelpunkt jedes biologischen Prozesses. Sie katalysieren als Enzyme einen komplexen Ablauf biochemischer Reaktionen, die in ihrer Gesamtheit ,,das Leben`` ausmachen. Um die molekularen Mechanismen der enzymkatalysierten Reaktionen zu verstehen und beispielsweise in der Medikamentenentwicklung (drug design) in sie eingreifen zu können, ist es notwendig, die 3D-Struktur der beteiligten Proteine zu kennen.
Die experimentelle Bestimmung dieser Struktur ist jedoch wesentlich zeitaufwendiger und teurer als die Bestimmung von Proteinsequenzen: Verdeutlicht wird dies durch die exponentiell wachsende Menge an bekannten Sequenzen im Vergleich zu der wesentlich langsamer wachsenden Anzahl an aufgeklärten Strukturen. Daher ist die Vorhersage der räumlichen Struktur, ausgehend von einer Protein-Sequenz, eines der zentralen Probleme der Bioinformatik.
Die Strukturvorhersage funktioniert derzeit allenfalls zufriedenstellend, wenn man die Struktur eines homologen Proteins, d.h. eines Proteins mit gleicher Abstammung, kennt. Dann kann man davon ausgehen, dass eine ähnliche 3D-Struktur vorliegt, und die bekannte Struktur kann als Vorlage (Template) benutzt werden, um mittels Homology Modelling ein Modell für die 3D-Struktur des anderen Proteins zu erstellen.
Im Rahmen einer Kooperation der Arbeitsgruppe Schomburg, Institut für Biochemie, und der Arbeitsgruppe Faigle/Schrader, ZAIK, wird daher versucht, das Auffinden entfernt homologer Proteine zu verbessern.
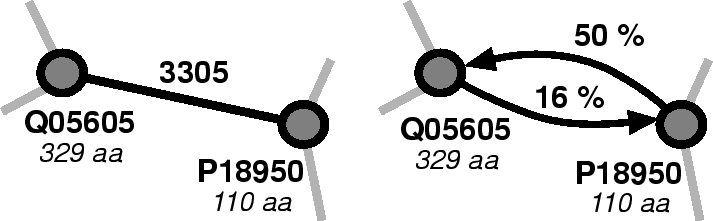
Das Prinzip des Ansatzes beruht auf der allgemein akzeptierten Annahme, dass zwei Sequenzen mit ausreichend hoher Sequenzähnlichkeit auch eine ähnliche Struktur besitzen. Für die Qualität der Sequenz-Struktur-Zuordnung ist die Sequenzidentität ein entscheidendes Maß. Aber selbst bei geringer Sequenzähnlichkeit können zwei Proteine homolog sein. Eine Eigenschaft der Homologie ist ihre Transitivität: wenn A und B sowie B und C sich aus dem gleichen Vorfahren ableiten, muss A auch einen Vorfahren mit C gemeinsam haben. Die Transitivität der Homologie wird in diesem Projekt genutzt, um auch entfernt homologe Proteine mit niedriger Sequenzidentität zu finden.
Da Heuristiken zum Sequenzvergleich, wie z.B. Blast und FASTA, bei geringer Sequenzähnlichkeit große Fehler produzieren, haben wir uns entschieden, den rechenaufwendigen Algorithmus nach Smith-Waterman zu verwenden. Für die 86.654 Proteinsequenzen in SwissProt, Release 39, ergibt sich damit ein Gesamtrechenaufwand von über 1.000 CPU-Tagen (UltraSparc CPU). Da das Problem jedoch perfekt zu verteilen ist, konnten wir sämtliche Workstations (ca. 25 CPUs) der Arbeitsgruppe als ein Rechencluster benutzen, und so über einen Zeitrahmen von 18 Wochen die Berechnung durchführen.
|
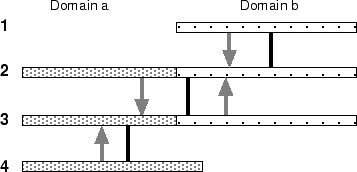
Die Ergebnisse der Sequenzvergleiche werden als Eingabe für ein graphenbasiertes Cluster-Verfahren benutzt. Ziel ist dabei herauszufinden, ob sich mittels der resultierenden Cluster tatsächlich mehr entfernt homologe Sequenzen auffinden lassen als bisher. Dies wird durch den Vergleich mit bestehenden Strukturdatensätzen untersucht. Dabei stellen sog. Multidomänenproteine ein Problem dar, dem wir durch Übergang zu einem gerichteten Graphen begegnen konnten.
 |
Die erste Phase dieses Projekts wurde im Rahmen einer gemeinsam von der Arbeitsgruppe Schomburg und Arbeitsgruppe Faigle/Schrader betreuten Diplomarbeit (Eva Bolten, ,,Eine graphenbasierte Clustermethode zur Detektion entfernt homologer Proteinsequenzen``) durchgeführt. Der Ansatz wird durch die Verwendung der gefundenen Cluster als Trainingsmenge für Profile-basierte Verfahren zum Finden entfernt homologer Sequenzen im Rahmen einer zweiten gemeinsam betreuten Diplomarbeit ausgebaut. Weiterhin soll der Zugriff auf die Cluster über eine entsprechende Weboberfläche ermöglicht werden.