 |
Hidden-Markov-Modelle (HMM) sind eine Klasse von stochastischen Modellen, die schon für eine Vielzahl von Anwendungsgebieten, von der Spracherkennung bis zur Simulation biochemischer Prozesse in Zellmembranen, erfolgreich eingesetzt wurden. Am ZAIK werden z.B. HMM auch für die Analyse und Simulation ökonomischer Zeitreihen, genauer von Bausparverträgen, benutzt.
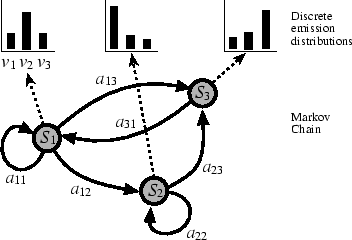
HMM bestehen aus einer Markov-Kette, also einer ,,gedächtnislosen`` Folge von Zufallsvariablen, den sogenannten Zuständen, die aber nicht direkt zu beobachten sind. Weiterhin gibt es für jeden dieser Zustände eine diskrete oder kontinuierliche Verteilung, anhand derer dann die beobachtbaren Ausgaben erzeugt werden. Ein HMM impliziert eine Wahrscheinlichkeitsverteilung auf der Menge aller möglichen Sequenzen von Ausgaben.
Für DNA-Sequenzen, also Abfolgen der Buchstaben A, C, G und T, kann in einem einfachen Beispiel ein Zustand des HMM jeweils einer funktional ausgezeichneten Region des DNA-Moleküls entsprechen (coding vs. non-coding). Diese unsichtbaren Zustände zeichnen sich z.B. durch unterschiedliche Sequenzkompositionen, also Unterschiede in der beobachteten relativen Häufigkeit der einzelnen Buchstaben A, C, G und T zwischen den verschiedenen Regionen aus. Ein mit entsprechendem Datenmaterial trainiertes HMM, kann dann z.B. für die Erkennung von kodierenden Regionen benutzt werden.
|
Weitere Anwendungsfelder von HMM in der Bioinformatik sind das Finden von Genen, Strukturvorhersagen, Verfahren zur Motivsuche, Finden entfernt homologer Proteinsequenzen, Multiple Alignments, Klassifizierung von DNA/Proteinsequenzen (Transkriptionseinheiten) und Modellierung von Proteinfamilien.
In der Literatur zu HMM, insbesonders in dem bekannten Tutorial-Artikel von Rabiner, werden drei Probleme in Bezug auf HMM erwähnt:
Da die von Rabiner betrachteten Anwendungen in der Spracherkennung auf natürliche Art und Weise ein geeignetes HMM nahelegten, ist es verständlich, dass eine viel grundlegendere Frage nicht gestellt wurde. Wie wählt man die Struktur oder Topologie eines HMM, also die Anzahl der Zustände und der zwischen ihnen erlaubten bzw. verbotenen Übergänge?
Die Frage ist aus zweierlei Gründen von Bedeutung. Zum einem ist die Anzahl der zu trainierenden Parameter, vereinfacht gesprochen, quadratisch in der Anzahl der Zustände. Bei einer fest vorgegebenen, beschränkten Datenmenge, wie es in der Praxis fast immer der Fall ist, ergeben sich damit Trainingsfehler für die Parameter, die mit der Modellgröße zunehmen. Des weiteren läuft man mit einer zu großen Anzahl an Parametern Gefahr, das Modell ,,überzutrainieren``, also Generalisierungsfähigkeit zu verlieren. Zum anderen geht die Anzahl der Zustände auch quadratisch in die Laufzeit der Algorithmen ein, die z.B. für die Datenbanksuche nach verwandten Proteinen benutzt werden.
Sofern das Anwendungsproblem nicht klare Vorgaben lieferte, wurden bisher weitgehend adhoc Verfahren, z.B. die sogenannte Model-Surgery, zur Auswahl einer geeigneten Topologie verwendet. Im Rahmen einer Promotion wurde am ZAIK ein Bayes'scher Ansatz entwickelt, der nach Vorgabe einer Prior-Distribution, die als erwartete Fehlerverteilung der Daten zu interpretieren ist, auf der Basis eines Clusterverfahrens Topologie und Modellparameter lernt.
 |
Mit diesem Verfahren können, ohne Bias für bestehende Problemstellungen, Modelle mit einer geringeren Anzahl an Zuständen und einer größeren Generalisierungsfähigkeit gelernt werden, die darüberhinaus für die typischen Bioinformatik-Anwendungen einen klaren Laufzeitvorteil bieten.