 |
Das Problem erfordert die Bestimmung der Schmelztemperatur jeder Zielsequenz mit jedem invers-komplementären Teilstück (als Probe). Dabei ist denkbar, dass die beiden nicht linear hybridisieren, sondern einzelne Basen ausgelassen werden und Schleifen oder andere Sekundärstrukturformen bilden. Die Berechnung kann mittels dynamischer Programmierung erfolgen, ähnlich wie in dem bekannten Algorithmus von Needleman-Wunsch bzw. Smith-Waterman, wobei jedoch anstatt der dort üblicherweise verwandten Kostenfunktion hier thermodynamische Parameter verwandt werden. Der Rechenaufwand ist allerdings enorm.
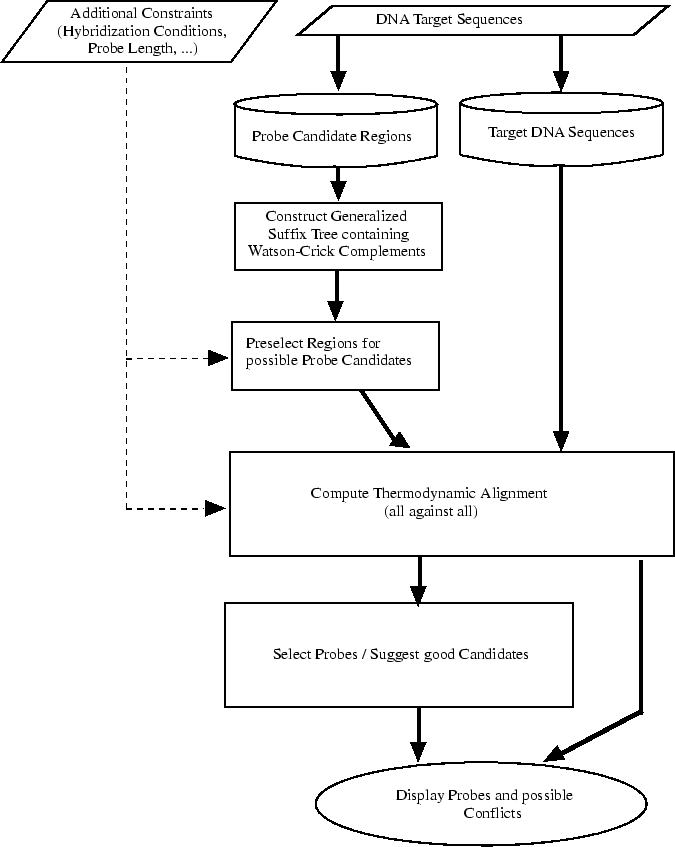
Durch eine Filterung ungeeigneter Teilsequenzen vor Beginn der eigentlichen Rechnungen kann bereits im Vorfeld eine dramatische Reduktion des Rechenaufwandes erreicht werden. Filterkriterien sind z.B. die Eindeutigkeit des gewählten Teilstückes, aber auch die Schmelztemperatur des zugehörigen Duplexes sowie die Länge der Probe. Erst im Anschluss daran beginnt die Berechnung aller möglichen Reaktionen. Abbildung zeigt schematisch den Ablauf dieses Prozesses.
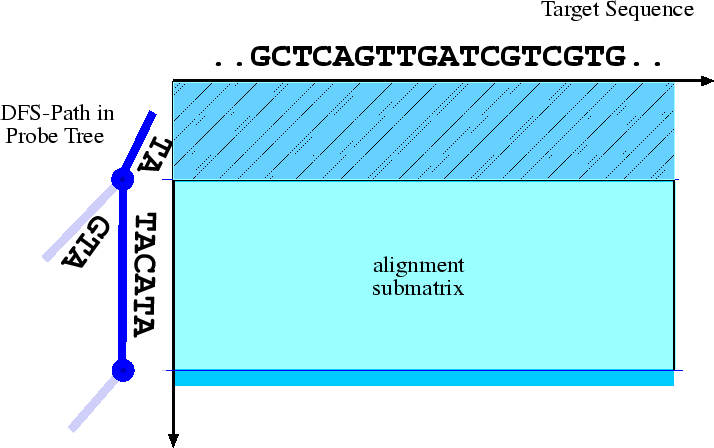
Genomische Sequenzen enthalten viele repetitive Motive. Dadurch kommt es relativ häufig vor, dass Proben gemeinsame Präfixe haben. Da alle Proben gegen die gleichen Zielsequenzen aligniert werden müssen, kann viel Zeit gespart werden, wenn dann solche Zwischenergebnisse gespeichert werden und bei ihrem erneuten Auftreten nicht neu berechnet werden müssen.
Eine geeignete Datenstruktur zum Erkennen solch gemeinsamer Präfixe
sind Suffixbäume. Ein Suffixbaum repräsentiert sehr
kompakt alle Teilsequenzen eines Strings. Dabei sind
die Kanten des Baumes mit Teilsequenzen beschriftet, und der Weg von
der Wurzel zum ![]() -ten Blatt des Baumes liefert genau den
-ten Blatt des Baumes liefert genau den ![]() -ten
Suffix der ursprünglichen Sequenz. Gemeinsame Präfixe von
Suffixen zeichnen sich durch einen gemeinsamen Pfad von der Wurzel aus.
Suffixbäume können kanonisch
so generalisiert werden, dass sie gleichzeitig die Suffixe mehrerer
Sequenzen enthalten.
-ten
Suffix der ursprünglichen Sequenz. Gemeinsame Präfixe von
Suffixen zeichnen sich durch einen gemeinsamen Pfad von der Wurzel aus.
Suffixbäume können kanonisch
so generalisiert werden, dass sie gleichzeitig die Suffixe mehrerer
Sequenzen enthalten.
Die Idee des am ZAIK entwickelten Verfahrens besteht nun darin, einen generalisierten Suffixbaum aus den Zielsequenzen aufzubauen. Dieser wird zunächst anhand oben genannter Kriterien gefiltert, um ungeeignete Proben auszuschliessen. Anschliessend wird der Baum in Tiefensuche durchlaufen, und für alle Knoten wird ein Alignment mit den Zielsequenzen berechnet. Dabei müssen Alignments für gemeinsame Präfixe nur einmal berechnet werden.
Aus den so berechneten Schmelztemperaturen kann anschließend mit Hilfe von im wesentlichen einfachen Sortierverfahren eine Probenmenge für den Chip ausgewählt werden. Dabei ist es möglich, sowohl im Pre-Processing, d.h. bei der Auswahl von Kandidaten, als auch bei der abschliessenden Auswahl Proben zu bestimmen, die sensitiv für eine ganze Familie von DNA-Sequenzen sind.